CO oxidation on Pt nanoclusters, size
and coverage effects
A density functional theory study
 Metal nanoclusters are widely used in industrial
catalysis, fuel cells, sensor devices, and have a great potential in various
bio-applications. For this reason, it is important to understand the mechanisms
and elementary steps of chemical reactions on the clusters. In this work CO
oxidation on Pt nanoclusters of approximately 1 nm in size was studied using
density functional theory (DFT). Reaction barriers on various sites of a
cuboctahedral 55‑atom cluster and of several two-layer plane clusters
representing (111) and (100) facets of the 147‑atom cluster have been
calculated at various coverage. In a discussion of chemical reactions on
clusters, the surface of the cluster can be considered as a combination of
small pieces of (111) and (100) terraces of a single crystal surface which form
facets of the cluster. Edges of the cluster can be seen as steps on the
surface, and corner sites as kinks.
Metal nanoclusters are widely used in industrial
catalysis, fuel cells, sensor devices, and have a great potential in various
bio-applications. For this reason, it is important to understand the mechanisms
and elementary steps of chemical reactions on the clusters. In this work CO
oxidation on Pt nanoclusters of approximately 1 nm in size was studied using
density functional theory (DFT). Reaction barriers on various sites of a
cuboctahedral 55‑atom cluster and of several two-layer plane clusters
representing (111) and (100) facets of the 147‑atom cluster have been
calculated at various coverage. In a discussion of chemical reactions on
clusters, the surface of the cluster can be considered as a combination of
small pieces of (111) and (100) terraces of a single crystal surface which form
facets of the cluster. Edges of the cluster can be seen as steps on the
surface, and corner sites as kinks.
CO catalytic oxidation on
platinum is believed to occur through the Langmuir‑Hinshelwood mechanism
between adsorbed CO and atomic oxygen O formed in O2 dissociations. Steps are known to provide stronger
adsorption and facilitate CO oxidation on single crystal surfaces[Yates]. DFT
calculations show that on clusters, CO molecules preferentially
adsorb on the corner sites of the cluster, while O-atoms can adsorb on both
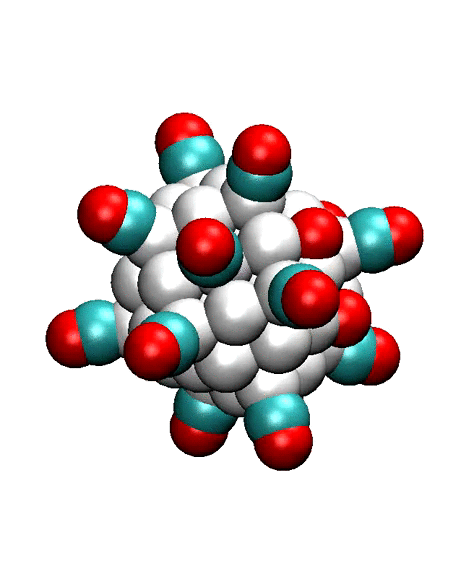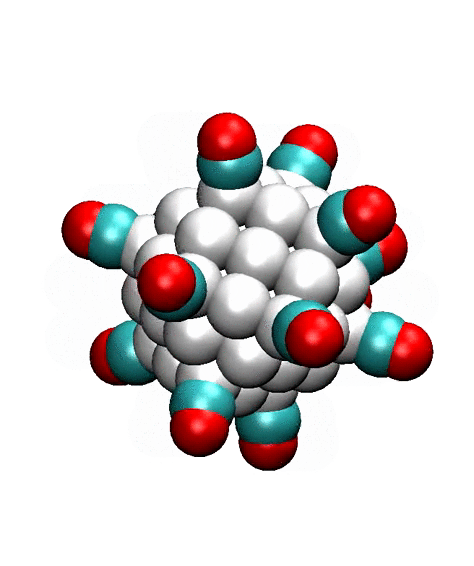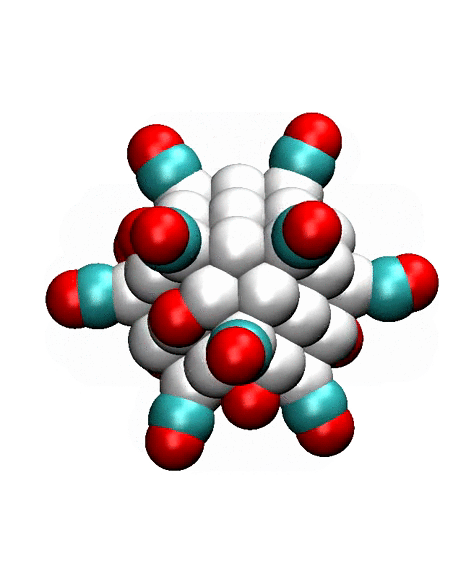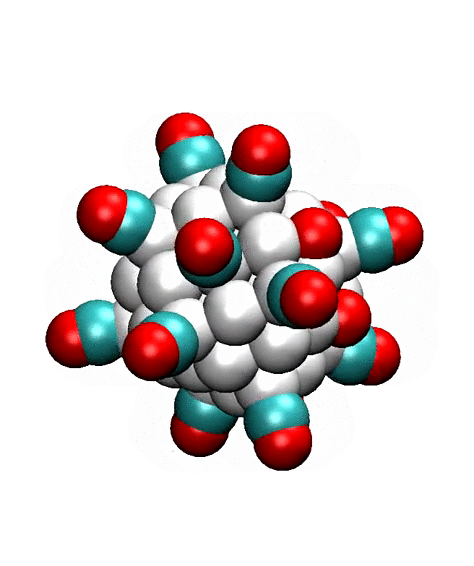
edges and facets, depending upon the coverage. One of stable high coverage
adsorption configurations is shown in Fig. 2.
From this low-energy configuration CO-molecules and O-atoms
can migrate and to react with each other on various sites of the
cluster to form a CO2 molecule. The newly formed CO2
molecule is weakly bound to the surface and desorbs immediately after the
formation.
Figure
2. Possible high-coverage adsorption configuration of CO and O-atoms on the
55-atom cuboctahedral Pt cluster
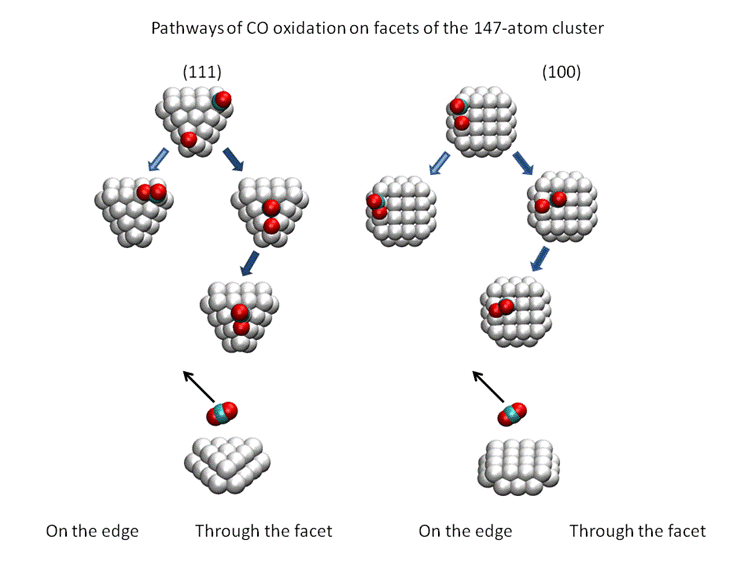 Two alternative pathways on the facets of the 147-atom cuboctahedral
cluster are shown in Fig. 3. One possibility is that adsorbed CO and O-atom
react on the edge (left columns), while another possibility is that CO molecule
migrates to an adsorption site on the facet, which is surrounded by the edges,
and then reacts (right column). The DFT calculations [Dobrin] show that
transition state is lower in the second case, especially at high coverage, when
other adsorbed species are present on the surface. This suggests that
‘surrounded’ adsorption sites on the facets may accelerate the CO oxidation.
Such sites are not present on the (111) facet of the 55-atom cluster, where
only 3-fold adsorption is possible. For this reason the 55-atom cluster can be
less efficient ion CO oxidation than 147-atom cluster.
Two alternative pathways on the facets of the 147-atom cuboctahedral
cluster are shown in Fig. 3. One possibility is that adsorbed CO and O-atom
react on the edge (left columns), while another possibility is that CO molecule
migrates to an adsorption site on the facet, which is surrounded by the edges,
and then reacts (right column). The DFT calculations [Dobrin] show that
transition state is lower in the second case, especially at high coverage, when
other adsorbed species are present on the surface. This suggests that
‘surrounded’ adsorption sites on the facets may accelerate the CO oxidation.
Such sites are not present on the (111) facet of the 55-atom cluster, where
only 3-fold adsorption is possible. For this reason the 55-atom cluster can be
less efficient ion CO oxidation than 147-atom cluster.
This result allows one qualitatively understand the
size-dependence of CO oxidation on platinum nanoclusters. Pt nanoclusters
provide the best conditions for CO oxidation if they are between 1 and 2 nm in
size, as it has been observed experimentally.[Haruta] This makes Pt clusters
different from gold nanoclusters, were CO oxidation was suggested to occur on
the corner sites and where small nanoclusters are always more efficient in CO
oxidation than larger ones. [Norskov]
Figure 3
[Yates] J. Xu, P. Henriksen and J. T. Yates, Jr., J. Chem. Phys., 1992, 97, 5250
[Dobrin]
S. Dobrin, Phys. Chem. Chem. Phys., 14, (2012) 12122 (pdf)
[Haruta] G. R.
Bamwenda, S. Tsubota, T. Nakamura, and M. Haruta, Catal. Lett., 1997,
44, 83.